Peptide identification from fragment spectra#
The peptide identification workflow is the cornerstone of data-dependent acquisition (DDA) quantification methods such as LFQ or TMT. To identify proteins by mass spectrometry, the proteins of interest in the sample are digested into peptides using a proteolytic enzyme (e.g., trypsin). The complex peptide mixture is then separated by liquid chromatography which is coupled to the mass spectrometer.
In DDA mode, the mass spectrometer first records the mass/charge (m/z) of each peptide ion and then selects the peptide ions individually for fragmentation to obtain sequence information via MS/MS spectra (Fig. 1). As a result for each sample, millions of MS and corresponding MS/MS are obtained which correspond to all peptides in the mixture.
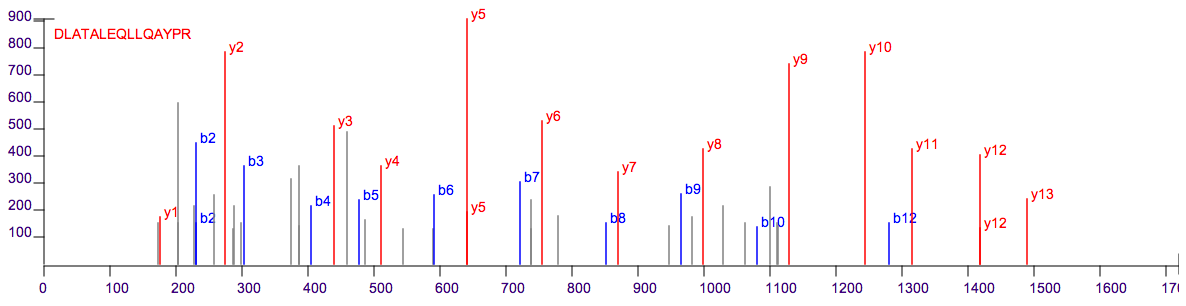
Fig. 1 MS/MS spectrum.#
In order to identify the MS/MS spectra, several computational algorithms and tools can now be used to identify peptides and proteins. The most popular ones are based on protein sequence databases, where the experimental MS/MS is compared with the theoretical MS/MS of each peptide obtained from the in silico digestion of the protein database (as reviewed in [RIVEROL2014]).
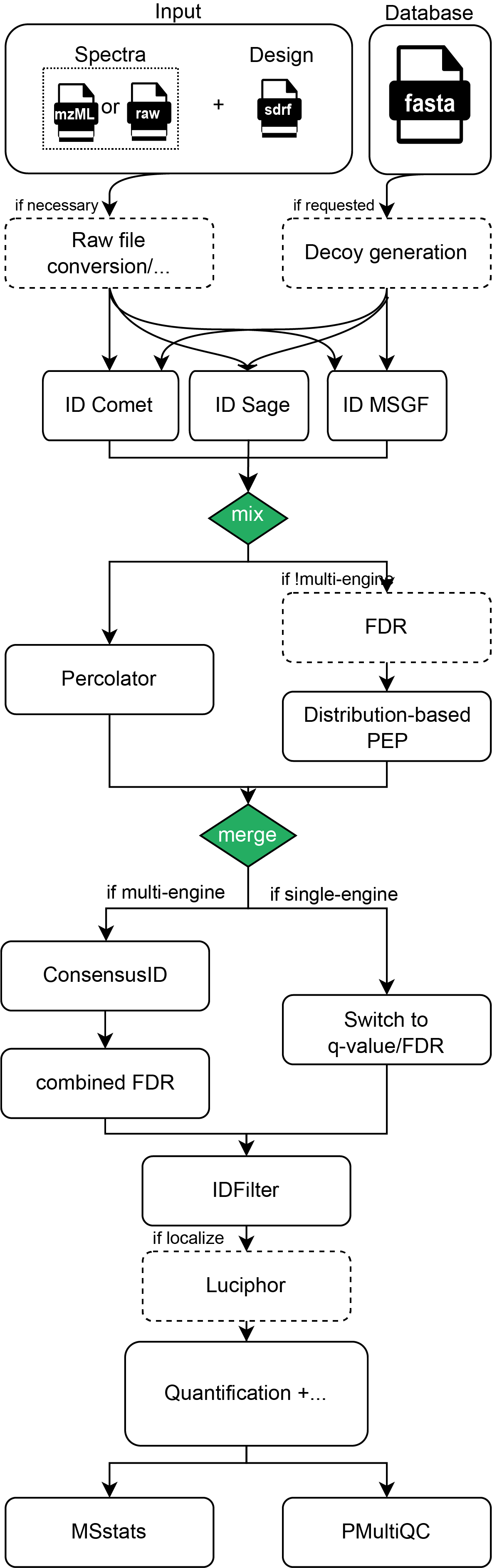
Peptide Identification#
The peptide identification step in the quantms pipeline can be performed (independently or combined) with two
different open-source tools : Comet search engine, MS-GF+ peptide search engine or Sage search engine. The parameters for the search engines Comet, MS-GF+ or Sage are
read from the SDRF input parameters including the post-translation modifications (annotated with UNIMOD accessions),
precursor and fragment ion mass tolerances, etc. The only parameter that MUST be provided by commandline to the
quantms workflow is the psm and peptide FDR threshold psm_pep_fdr_cutoff (default value 0.01).
Note
Using multiple database search engine combined can yield up to 15% more peptides compared to using only one search engine. However, you need to be aware that adding another search engine will increase the CPU computing time. Benchmarks.
When multiple search engines are used `--search_engines msgf,comet` the results for each input file are
combined into one single identification file including the combination of all listed search engines.
To bring scores from different search engines to a comparable level, the posterior (error) probability output
from either Percolator or the distribution-fitting approach is used.
The OpenMS tool ConsensusID is used to combine the results from different search engines [NAHNSEN2011]. ConsensusID provides multiple algorithms to combine PSMs from multiple search engines. The default option and more widely tested is best. In this option, the best chooses the PSM with the highest probability. Here it is enough for each engine to provide only one top hit.
Note
The other two more advanced algorithms are: PEPMatrix - calculates a matrix of similarities across sequences of different engines to increase the weight for sequences that have a similar counterpart for another engine. PEPIons - calculates a matrix of the number of shared matched ions across sequences of different engines to increase the weight for sequences that have a similar counterpart for another engine.
Re-scoring of peptide-spectrum matches#
To bring scores on a comparable level and to potentially improve their ranking the pipeline employs PSM re-scoring procedures. Re-scoring methods used multiple properties from the PMS’s like retention time, to increase the differences between the target and decoy peptides. Multiple algorithms and tools has been developed in proteomics to boost the number of peptide identifications and the score of the final peptide hits.
quantms provides two different algorithms and tools for re-scoring of the peptide identifications: Percolator (SVM-based re-scoring) and Mixture-model-based re-scoring using probability distributions. By default, quantms uses the Percolator (SVM-based re-scoring) algorithm, which has proved to increase peptide identifications for Comet search engine and MS-GF+ peptide search engine search engines.
Note
In some cases, Percolator fails to boost the original search engines, especially in cases like small datasets where the number of peptide identifications is insufficient). As an alternative, quantms offers a fully parameterized unsupervised or semi-supervised distribution approach inspired by PeptideProphet. The distribution families are currently fixed (Gumbel distribution for incorrect and Gaussian for correct PSMs). In order to switch the use to idpep, the user should provide the following parameter: –posterior_probabilities ‘fit_distributions’
After the re-scoring of the peptide identification and combination of the results from multiple search engines (if multiple search engines are allowed), quantms allows the fdr calculation steps (read more in False discovery rate estimation) and optionally the modification sites localization (read more in Modification localization).
References#
Perez-Riverol Y, Wang R, Hermjakob H, Müller M, Vesada V, Vizcaíno JA. Open source libraries and frameworks for mass spectrometry based proteomics: a developer’s perspective. Biochim Biophys Acta. 2014 Jan;1844(1 Pt A):63-76. doi: 10.1016/j.bbapap.2013.02.032. Epub 2013 Mar 1. PMID: 23467006; PMCID: PMC3898926.
Nahnsen S, Bertsch A, Rahnenführer J, Nordheim A, Kohlbacher O. Probabilistic consensus scoring improves tandem mass spectrometry peptide identification. J Proteome Res. 2011 Aug 5;10(8):3332-43. doi: 10.1021/pr2002879. Epub 2011 Jun 23. PMID: 21644507.